

Le parlement européen renforce les procédures de conformité des dispositifs médicaux
Un an après avoir été promulgué, le règlement européen relatif aux dispositifs médicaux est entré en vigueur le 26 mai 2021. Cécile Vaugelade (SNITEM) a fait une synthèse des nouvelles contraintes qui pèseront sur les industriels du secteur au cours d’un webinaire dédié.
Une refonte totale de la règlementation européenne en matière de dispositifs médicaux (DM) a été mise en place le 26 mai 2021, au travers de la mise en application du nouveau règlement UE 2020/561 du Parlement européen et du conseil du 23 avril 2020. Ce règlement, qui a fait l’objet d’un report d’un an pour cause de crise sanitaire, renforce significativement les contraintes pour l’obtention du marquage CE médical.
Un marquage CE qui concerne la totalité des dispositifs médicaux
À l’occasion d’un webinaire organisé le 26 mai 2021, Cécile Vaugelade, Directrice des affaires technico-réglementaire au sein du Syndicat National de 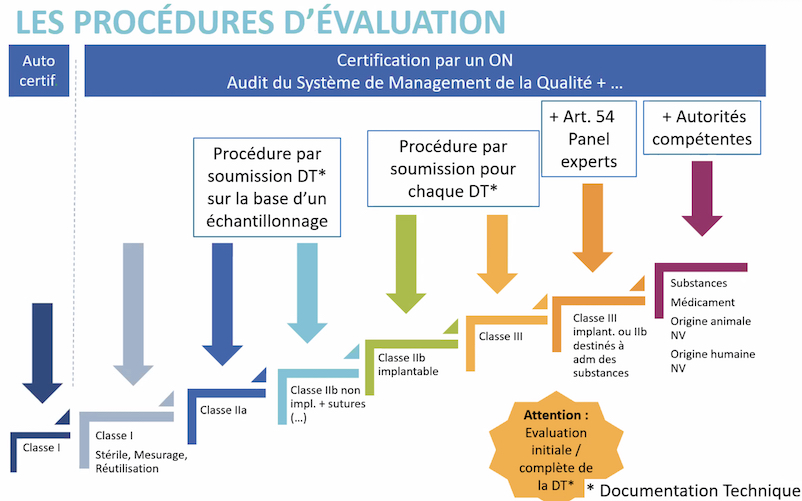 l’Industrie des Technologies Médicales (SNITEM), a fait une synthèse de ce nouveau texte et mis en lumière les mesures supplémentaires qui devront être appliquées pour améliorer le processus de mise sur le marché des DM. Les différentes étapes qui conduisent à la commercialisation de ces matériels s’apparentait déjà à un parcours du combattant. Mais elles sont désormais renforcées, ajoutant encore des contraintes aux fabricants et distributeurs.
l’Industrie des Technologies Médicales (SNITEM), a fait une synthèse de ce nouveau texte et mis en lumière les mesures supplémentaires qui devront être appliquées pour améliorer le processus de mise sur le marché des DM. Les différentes étapes qui conduisent à la commercialisation de ces matériels s’apparentait déjà à un parcours du combattant. Mais elles sont désormais renforcées, ajoutant encore des contraintes aux fabricants et distributeurs.
Alors, quelles sont ces nouvelles dispositions qui sécurisent la qualité de ces DM ? La première est l’élargissement du domaine du marquage CE, qui prend en compte désormais tous les DM, même ceux qui comportent un faible risque pour le patient – à l’exclusion des probiotiques -, y compris les implants à visée esthétique. D’autre part, certains DM voient leur classification modifiée à la hausse. C’est le cas des logiciels métiers ou certains implants rachidiens ou articulaires qui passent en classe III.
Des niveaux d'exigence revus à la hausse
Les niveaux d’exigence dans le processus d’évaluation sont également revus à la hausse, avec une évaluation clinique renforcée par des essais cliniques et de nouvelles études formalisées. Quant aux organismes notifiés (ON), qui ont pour mission d’évaluer les dossiers de conformité des DM, ils devront certifier des compétences de leurs personnels évaluateurs et devront se conformer à de nouvelles méthodes d’évaluation plus complètes, un panel d’experts étant exigé pour évaluer la conformité des DM de classe III par exemple.
La chaîne de distribution est également impactée par le nouveau texte, notamment le transport et le stockage, de même que la transparence et la traçabilité des produits. La base de données EUDAMED répertoriera les identifiants uniques des dispositifs (UDI) et les dispositifs implantables feront l’objet d’une carte d’implant au niveau européen, transposant ce qui se fait déjà en France. Les informations contenues dans cette base seront accessibles aux patients et aux professionnels de Santé qui feront l’usage de ces DM.
Ces nouvelles mesures, sensées harmoniser les pratiques au niveau européen, se doivent toutefois de s’adapter aux réglementations nationales. En France, la Loi Bioéthique apportera éventuellement quelques ajustements qui feront l’objet d’ordonnances.
SUR LE MÊME THÈME

Les principes de radiobiologie et de radioprotection synthétisés dans le Modern Radiology ebook
Dans la série de documents pédagogiques Modern Radiology ebook qu’elle édite depuis quelques années à l’attention des professionnels de la spécialité, l’European Society of Radiology a publié un recueil de Principes de radiobiologie et de radioprotection.
11/03/2026 -

Cybersécurité : une stratégie gouvernementale ambitieuse
Les menaces de cybersécurité, qui intéressent fortement les acteurs de l’imagerie médicale, seront combattues, durant les quatre prochaines années, par une stratégie gouvernementale. Celle-ci vise notamment à renforcer la résilience cyber et garder la maîtrise des fondements numériques, à travers un...
09/02/2026 -

Une infirmière en IRM ou l'exercice illégal de la profession de MERM
Une annonce émanant d’un grand groupe de radiologues lance la recherche d’une infirmière pour ses installations d’IRM afin qu’elle assure une gestion du patient alors que le MERM pilote la modalité dans un Cockpit. Mais cette annonce va trop loin et promeut un exercice illégal du métier de MERM. La...
26/01/2026 -

La communauté radiologique vent debout contre les dispositions réglementaires qui dévalorisent les actes de radiologie
Tous les radiologues, hospitaliers et libéraux, une fois n’est pas coutume, ont collaboré pour diffuser, le 17 Novembre 2025, un communiqué alertant sur les dangers que la spécialité encourt si les nouvelles dispositions réglementaires relatives à la dévalorisation des actes d’imagerie médicale.
19/11/2025 -


L'ASNR cible les pratiques de la téléradiologie sur le champ de la radioprotection
Dans une étude qu’elle vient de publier, l’ASNR pointe des points de fragilité pour la radioprotection dans le domaine de la téléradiologie. Communication altérée entre téléradiologues, MERM et prescripteurs sont pointés du doigt ainsi qu’une et surcharge de travail pour les paramédicaux. Elle édite...
06/11/2025 -

Les nouveaux tarifs des actes d'imagerie sont publiés
La Décision du 14 octobre 2025 relative à la liste des actes d'imagerie fait craindre des fermetures de cabinets radiologie libérale, selon la FNMR. Ce texte comprend des réductions de valorisation d’actes de radiologie diagnostique et interventionnelle mais c'est l’imagerie en coupe qui subit les p...
21/10/2025 -

La Loi de finances de la sécurité sociale cible l'imagerie lourde et la radiothérapie libérale
Le Projet de loi de financement de la sécurité sociale 2026 vient donc de mettre en pratique les annonces faites l’été dernier par l’IGAS et de l’IGS. Les forfaits techniques d’imagerie lourde et la radiothérapie libérale sont en point de mire pour enclencher des économies à l’assurance maladie. La...
18/10/2025 -

LETTRE D'INFORMATION
Ne manquez aucune actualité en imagerie médicale et radiologie !
Inscrivez-vous à notre lettre d’information hebdomadaire pour recevoir les dernières actualités, agendas de congrès, et restez informé des avancées et innovations dans le domaine.